












 will be held in Guangzhou, Shanghai, Chengdu and Beijing")








Mapping the ‘chromatinome’: New integrative functional genomics platform for profiling genome regulation with a massively-parallel screen of chromatin modifiers.
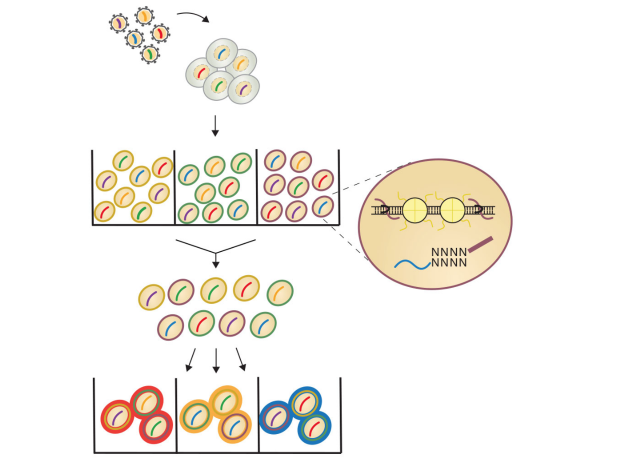
In a new resource for the scientific community, published today in Nature Biotechnology, researchers in the lab of Neville Sanjana at the New York Genome Center (NYGC) and New York University (NYU) developed CRISPR-sciATAC, a novel integrative genetic screening platform that jointly captures CRISPR gene perturbations and single-cell chromatin accessibility genome-wide. With this technology, they profile changes in genome organization and create a large-scale atlas of how loss of individual chromatin-altering enzymes impacts the human genome. The new method harnesses the programmability of the gene editing system CRISPR to knock-out nearly all chromatin-related genes in parallel, offering researchers deeper insights into the role of DNA accessibility in cancer and in rare diseases involving chromatin.
Recent advances in single-cell technologies have given scientists the ability to profile chromatin, the complex of DNA and proteins that resides within the nucleus of individual cells. Chromatin is often called the “gatekeeper” of the genome because its proteins act as packaging elements for the DNA, either promoting or refusing access to it. This controls gene expression processes in the cell, such as turning on or off specific genes. Changes in the chromatin landscape have been linked to diverse human traits and diseases, most notably cancer.
In an initial demonstration of CRISPR-sciATAC, the Sanjana Lab team designed a CRISPR library to target 20 chromatin-modifying genes that are commonly mutated in different cancers, including breast, colon, lung and brain cancers. Many of these enzymes act as tumor suppressors and their loss results in global changes in chromatin accessibility. For example, the group showed that loss of the gene EZH2, which encodes a histone methytransferase, resulted in an increase in gene expression across several previously silenced developmental genes.
“The scale of CRISPR-sciATAC makes this dataset very unique. Here, in a uniform genetic background, we have accessibility data capturing the impact of every chromatin-related gene. This provides a detailed map between each gene and how its loss impacts genome organization with single-cell resolution,” said Noa Liscovitch-Brauer, a postdoctoral fellow in Sanjana’s lab at the New York Genome Center and NYU and the study’s co-first author.
In total, the team targeted more than 100 chromatin-related genes and developed a “chromatin atlas” that charts how the genome changes in response to loss of these proteins. The atlas shows that different subunits within each of the 17 chromatin remodeling complexes targeted can have different effects on genome accessibility. Surprisingly, nearly all of these complexes have subunits where loss triggers increased accessibility and other subunits with the opposite effect. Overall, the greatest disruption in transcription factor binding sites, which are important functional elements in the genome, was observed after loss of AT-rich interactive domain-containing protein 1A (ARID1A), a member of the BAF complex. Mutations in BAF complex proteins are estimated to be involved in 1 out of every 5 cancers.
In addition to the CRISPR-sciATAC method, the team also developed a suite of computational methods to map the dynamic movements of the nucleosomes, which are the protein clusters that DNA is wrapped around. When there are more nucleosomes, the DNA is tightly wound and less available to bind transcription factors. This is exactly what the team found at specifical transcription factor binding sites involved in cell proliferation after CRISPR knock-out of ARID1A. When targeting a different chromatin-modifying enzyme, these same sites underwent an expansion in nucleosome spacing, demonstrating the dynamics of nucleosome positioning at specific sites in the genome. The CRISPR-sciATAC method allowed the team to systematically explore this genome plasticity for multiple chromatin-modifying enzymes and transcription factor binding sites.
“We really focused on making CRISPR-sciATAC an accessible technique — we wanted it to be something that any lab could do. We produced most of the key enzymes in-house and used simple methods for single-cell isolation that do not require microfluidics or single-cell kits,” said Antonino Montalbano, a former postdoctoral fellow in Sanjana’s lab at the New York Genome Center and NYU and the study’s co-first author.
To develop the CRISPR-sciATAC technology, the researchers used a mix of human and mouse cells to create a tagging/identification process that allowed them to split and barcode the nuclei of cells as well as capture the single-guide RNAs required for CRISPR targeting. The work builds off prior single-cell combinatorial indexing ATAC-seq (sciATAC-seq) work from Dr. Jay Shendure at the University of Washington and other groups developing new single-cell genomics methods. CRISPR-sciATAC also uses a unique, easy-to purify transposase that was developed in the NYGC’s Innovation Technology Lab. A key technical hurdle was optimizing experimental conditions to simultaneously capture the CRISPR guide RNAs and genome fragments for accessibility profiling while also keeping the nuclear envelope of each cell intact.
“Integrating chromatin accessibility profiling into the genome-wide CRISPR screens provides a new lens for us to understand gene regulation,” said Sanjana, assistant professor of biology at NYU, assistant professor of neuroscience and physiology at NYU Grossman School of Medicine, core faculty member at NYGC, and the study’s senior author. “With CRISPR-sciATAC, we have a comprehensive view into how specific chromatin-modifying enzymes and complexes change accessibility and orchestrate the interactions that control gene expression. Chromatin sets the stage for gene expression, and here we can measure the impact of different mutations on chromatin rapidly. We hope this atlas will be a broadly useful resource for the community and that CRISPR-sciATAC will be used to produce similar atlases in other biological systems and disease contexts.”















